There are not many incentives for young neuroscientists to think aloud about big questions. Due to lack both of knowledge and authority, discussing very broad questions like how the brain works risks to be embarrassing at best. Still, I feel that not doing so is even more detrimental since it restrains the potential for internal development: Exposing one’s thoughts comes with the potential to refine them and to dissociate from them, thereby bringing down or advancing ideas that might have got stuck.
I want to make it a habit to report some of the thoughts about the brain that marked me most during the past twelve month at the end of each year, with the hope to advance and structure the progress in the part of my understanding of the brain which is not immediately reflected in journal publications. Other year-end write-ups: 2018, 2019, 2020, 2021, 2022, 2023, 2024.
How I got interested in dendrites
The lines of thought described in the following actually go back as far as to 2015. I was planning to switch from calcium imaging to whole-cell recordings as my main laboratory technique and started understanding the power of studies relying on this technique. In summer 2015, I came across a paper by Katie Bittner in Jeff Magee’s lab [1] (followed up by another paper [2]). Those papers showed that electrical “plateau potentials” can drive the formation of a place cell within a single trial. The authors established in vivo whole-cell recordings deep in the CA1 region of the hippocampus. Using this technique, they could generate plateau potentials by somatic current injection and thereby trigger the generation of a place field. As probably many others, I was immediately struck by this single-shot learning behavior, but, also due to lack of background knowledge, I was not yet able to see it in a larger context.
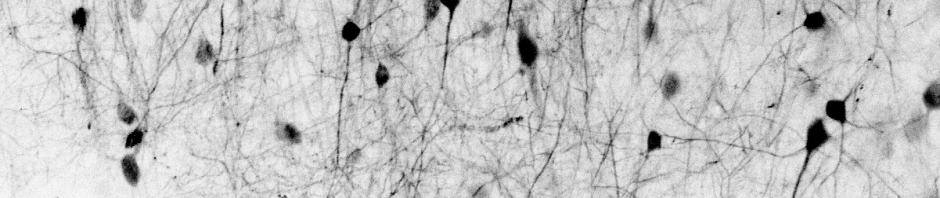
Later, when I was searching for potential postdoc positions in 2018, I first fully encountered the mystery of the apical dendrites of pyramidal neurons. Pyramidal neurons in layer 5 of the mammalian cortex grow from their soma a “basal” dendritic tree that remains rather local in layer 5, and in addition a thick “apical” trunk that goes up to layer 1, where it branches into many small apical dendritic processes (the apical “tuft”).
I was particularly intrigued by a review by Matthew Larkum from 2013 suggesting a specific function for the apical tuft of L5 neurons. This suggested function would be to detect almost coincident somatic activity and strong input to apical dendrites, resulting in a calcium spike in the apical trunk and leading to somatic bursting [3].
Problem 1: Top-down input to the apical dendrites of pyramidal neurons
Apical dendrites of L5 neurons are thought to receive mainly top-down input, whereas the basal dendrites are predominantly contacted by bottom-up input. For example, basal dendrites of the primary visual cortex would be expected to receive more sensory input from the primary thalamic region, whereas apical dendritic processes would receive rather context-related input from brain regions higher in the sensory hierarchy. I do not know how well this separation of top-down and bottom-up inputs for apical and basal dendrites holds true – in an earlier blog post I have described why I am generally not a fan of hierarchies like this top-down/bottom-up connectivity scheme, although I still find it a fascinating idea.
Since I’m currently working next door to the lab of Georg Keller, who is interested in predictive processing in visual cortex (check out his 2018 review [4]), I could not prevent myself from wondering whether this top-down contextual input to the apical dendrites could simply be predictions. This possibility is also mentioned in the Larkum review [3]. However, in the theory of predictive processing, predictions (here: apical input) should be subtracted from the sensory input (here: basal input), or the sensory input should be subtracted from the predictions. As mentioned above in the review by Matthew Larkum, however, the apical trunk seems to compute the coincidence of those inputs rather than the difference. Therefore, this somehow does not seem to make sense.
There are ideas how to implement predictive processing using L5 pyramidal cells nevertheless. For example, there is an interesting computational model that is pretty detailed (described by Sacramento et al. [5]). The idea here is that the apical compartment does not simply signal top-down input, but encodes an error signal between local inhibitory signals and top-down excitatory input. Some assumptions of this model seem to be unrealistic and many aspects of the model are simply unconstrained by experiments, but it is an interesting starting point nethertheless.
Problem 2: Coupling between apical dendrites and the soma
Overall, this leaves me with the impression that the apical compartment might be something crucial to understand. The separation of processing in apical and somatic compartments is an assumption that seems legit given the large electrotonic distance between soma and the apical dendrite. In addition, this assumption is supported by experimental data (e.g., Cichon & Gan, 2015 [6]; Seibt et al., 2017 [7]; and some other studies). However, for all of those studies, no direct evidence for the decoupling of somatic and apical activity was available. Direct evidence would mean simultaneous recording of somatic and dendritic activity, which is challenging even for an indirect method as calcium imaging due to the large spatial separation of soma and distal apical dendrite. Probing of calcium dynamics in a direct way so far has not shown strong decoupling of somatic and dendritic activity (e.g., Helmchen et al., 1997 [8]; or more recently Kerlin et al., 2018 [9]). To be more precise, these studies showed that almost all calcium events in the apical dendrites – with very few exceptions – were temporally coupled to backpropagating action potentials. This seems to be somehow at odds with the idea of separate processing in somatic and apical compartments.
Of course, this is only about dendritic calcium signals, not about the voltage. Recording of the voltages over multiple locations of a dendritic tree, for which there is currently no reliable method, could potentially result in a different picture. Plus, the brain areas and behavioral contexts are not immediately comparable between the behavioral tasks in these experiments. For example, Helmchen et al. [8] used anesthetized rats; Kerlin et al. [9] trained their mice extensively before experiments; Cichon et al. [6] recorded dendritic activity during a weird learning paradigm that might have resulted in a lot of confusion in the mice; and Seibt et al. [7] focused on dendritic activity in mice and rats during sleep.
As a result of these (seemingly) contradictory results, I’m intrigued by the unresolved question how tightly the activities of soma and apical dendrites of L5 neurons are indeed coupled. Or rather, under which circumstances both compartments become uncoupled. The answer to this question is completely unclear to me.
Problem 3: What do bursts of pyramidal neurons signal?
It is however clear that somatic action potentials to some extent invade the apical dendritic tree. This does not seem to be a random side effect, since it was reinforced by evolution by the insertion of active conductances into the dendritic tree. One possible purpose of this backpropagating action potential could be to activate the inputs of the apical dendrite, resulting in non-linear amplification in the distal dendrites or in the apical trunk (as described by the Larkum review [3]) and thereafter in somatic burst firing. What is the purpose of these bursts? I can come up with two possible explanations:
(1) As suggested by the experiments in the Magee lab ([1][2]), the bursts could be a strong intracellular signal to reinforce recently activated context synapses. – If so, in which synapses would plasticity occur, in basal or rather apical dendrites? In the studies from the Magee lab in the hippocampus plasticity in synapses of the stratum radiatum of CA1 was observed [2]; those synapses are thought to provide spatial context. How would this translate to cortex?
(2) A second possible function of bursts could be to signal not inside of a neuron, but between neurons. Regular spiking is ideally suited to drive postsynaptic neurons with depressing synapses, i.e., only the first spike of a rapid sequence of spikes would trigger substantial synaptic release of neurotransmitters. Bursting, however, is ideally suited to drive postsynaptic neurons that are connected via facilitating synapses. The bursts would therefore be a very sparse code that could signal a coincidence of somatic spiking and apical input to the downstream neuron. In a theoretical study, Naud & Sprekeler [10] investigated the potential of such multiplexing through simple spikes and bursts for separate processing of top-down and bottom-up input in a hierarchical network. And Blake Richards mentioned (in a talk that I’ve watched on youtube, start at min 22:14), while not going into the details, the possibility to use this multiplexing for helping to solve the “credit assignment problem”.
Brief digression: The credit assignment problem is about the question how a neuron somewhere in the brain network can learn to weigh the incoming information in order to become better at a given task. This problem is also addressed by the previously mentioned paper by Sacramento et al. [5], and there is a paper by Guerguiev et al. [11] that goes into a similar direction but is a bit chaotic. Lillicrap and Richards just published a review on how the credit assignment could be solved using (apical) dendrites [12]. It is important to note that both Sacramento et al. and Guerguiev et al. suggest solutions that are approximations of “backpropagation” of errors, the solution to the credit assignment problem that has been fameously found for artificial neural networks. Backpropagation, however, follows a linearized gradient in error space and therefore only allows for small synaptic changes – and thus does not really allow for the single-shot learning behavior that has been observed in animals ([1][2]). Therefore I’m not sure whether it is good idea to search for an implementation of an algorithm similar to backpropagation in the brain.
Problem 4: It’s getting ever more complex
In addition to the open questions mentioned above, some other points related to the function of apical dendrites are also not clear.
For example, what role do inhibitory neurons play that specifically target the apical dendrite? With a disinhibtory circuit motif, interneurons could specifically gate plasticity by blocking inhibition of an apical dendrite (check this review by Letzkus et al. [13]). Following this line of thought, it is, however, not clear to me whether disinhibition is (branch- or neuron-) specific or rather a broad, global gating mechanism of plasticity that allows for specific plasticity by other means.
As another example, it is to some extent clear how the membrane potential behaves in vivo in the soma – but less so in the dendrites of the very same neurons. Dendrites might integrate much fewer inputs than a soma and thereby exhibit much stronger voltage fluctuations – unless there is a precise local balance of excitatory and inhibitory inputs to a single dendrite (this question is based on work I did in zebrafish). A recent study addressed this question of balancedness partially by mapping the co-localization of excitatory and inhibitory neurons on a full tree of L2/3 pyramidal neurons [14].
In the context of balanced networks, I’m also wondering whether apical dendrites in living, unanesthetized brains operate in a high-conductance state as a result of strong excitatory and inhibitory inputs, which has been suggested for balanced networks. If so, I would be interested to know how the mass of open input channels in vivo would affect the coupling between dendritic segments compared to ex vivo slice studies. For this particular question, I’m not sure whether I’m just ignoring existing literature on the subject or whether these questions have simply not yet been addressed experimentally.
Summary
What happens in apical dendrites of L5 pyramidal neurons remains mysterious to me, in particular if the neuron is integrated in the cortex of a behaving animal or human being. I do not know which kind of events trigger activity of the apical dendrite, and I do not know under which circumstances and how the apical compartment communicates with the soma. I wonder how well the compartments are connected electrically in vivo. And it is an open question how both synaptic plasticity and non-plastic information processing are affected by activation of the apical trunk or the more distal apical tuft. Despite this lack of knowledge, I would guess that understanding apical dendrites is not sufficient, but probably necessary to understand what a cortical region does as a whole.
Maybe I’m wrong about some of my interpretations; probably I’m overlooking some import studies. If something is wrong in my guesses and interpretations, or if I am missing an important piece of experimental or theoretical evidence, please let me know. I do not have an agenda that I want to defend but instead would like to understand. Therefore, critical comments are even more welcome than positive feedback!
.
References
[1] Bittner KC, Grienberger C, Vaidya SP, Milstein AD, Macklin JJ, Suh J, Tonegawa S & Magee JC. Conjunctive input processing drives feature selectivity in hippocampal CA1 neurons. Nature Neuroscience (2015).
[2] Bittner KC, Milstein AD, Grienberger C, Romani S, & Magee JC. Behavioral time scale synaptic plasticity underlies CA1 place fields. Science (2017).
[3] Larkum ME. A cellular mechanism for cortical associations: an organizing principle for the cerebral cortex. Trends in Neurosciences (2013).
[4] Keller GB, & Mrsic-Flogel TD. Predictive Processing: A Canonical Cortical Computation. Neuron (2018).
[5] Sacramento J, Costa RP, Bengio Y, & Senn W. Dendritic cortical microcircuits approximate the backpropagation algorithm. Advances in Neural Information Processing Systems (2018).
[6] Cichon J, & Gan WB. Branch-specific dendritic Ca2+ spikes cause persistent synaptic plasticity. Nature (2015).
[7] Seibt J, Richard CJ, Sigl-Glöckner J, Takahashi N, Kaplan DI, Doron G, Limoges D, Bocklisch C, & Larkum ME. Cortical dendritic activity correlates with spindle-rich oscillations during sleep in rodents. Nature Communications (2017).
[8] Helmchen F, Svoboda K, Denk W, & Tank DW. In vivo dendritic calcium dynamics in deep-layer cortical pyramidal neurons. Nature Neuroscience (1999).
[9] Kerlin A, Mohar B, Flickinger D, MacLennan BJ, Davis C, Spruston N & Svoboda K. Functional clustering of dendritic activity during decision-making. bioRxiv (2018).
[10] Naud R, & Sprekeler H. Sparse bursts optimize information transmission in a multiplexed neural code. PNAS (2018).
[11] Guerguiev J, Lillicrap TP, & Richards, BA. Towards deep learning with segregated dendrites. eLife (2017).
[12] Richards BA, & Lillicrap TP. Dendritic solutions to the credit assignment problem. Current Opinion in Neurobiology (2019).
[13] Letzkus JJ, Wolff SB, & Lüthi A. Disinhibition, a circuit mechanism for associative learning and memory. Neuron (2015).
[14] Iascone DM, Li Y, Sumbul U, Doron M, Chen H, Andreu V, & Polleux F. Whole-neuron synaptic mapping reveals local balance between excitatory and inhibitory synapse organization. bioRxiv (2018).

 into a relationship that is rather linear in time,
into a relationship that is rather linear in time,  . Such that at time points close to the turnaround of the sine (blue time point below), the ‘black box lens’ would increase the angular deflection angle, eventually inversing the sine function:
. Such that at time points close to the turnaround of the sine (blue time point below), the ‘black box lens’ would increase the angular deflection angle, eventually inversing the sine function:
 from the center of the lens.
from the center of the lens.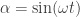 . The scanned beam hits the black box lens at a position
. The scanned beam hits the black box lens at a position 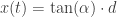 with the distance
with the distance 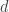 between the resonant scanner and the lens. The refractive power
between the resonant scanner and the lens. The refractive power 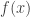 of the lens must therefore change with
of the lens must therefore change with 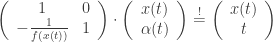
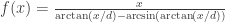
 and
and  that are influenced by each other:
that are influenced by each other:
 , the variable
, the variable