This is a blog post dedicated to those who start with calcium imaging and wonder why their live images seem to drown in shot noise. The short answer to this unspoken question: that’s normal.
Introduction
Two-photon calcium imaging is a cool method to record from neurons (or other cell types) while directly looking at the cells. However, almost everyone starting with their first recording is disappointed by the first light they see – because the images looked better, with more detail, crisper and brighter in Figure 1 of the lastest paper. What these papers typically show, is, however, not a snapshot of a single frame, but a carefully motion-corrected and, above all, averaged recording.
In reality, it is often not even necessary to see every structure in single frames. One can still make efficient use of such data that seemingly drown in noise, and you do not have to necessarily resort to deep learning-based denoising to make sense of the data. Moreover, if you can see your cells very clearly in a single frame, it is in many cases even likely that either the concentration of the calcium indicator or the applied laser power is too high (both extremes can induce damage and perturb the neurons).
To demonstrate the contrast between typical single frames before and beautiful images after averaging for presentations, here’s a gallery of recordings I made. On the left, a single imaging frame (often seemingly devoid of any visible structure). On the right, an averaged movie. (And, yes, please read this on a proper computer screen for the details, not on your smartphone.)
Hippocampal astrocytes in mice with GCaMP6s
Here, I imaged hippocampal astrocytes close to the pyramidal layer of hippocampal CA1. Laser power: 40 mW, FOV size: 600 µm, volumetric imaging rate: 10 Hz (3 planes), 10x Olympus objective. From our recent study on hippocampal astrocytes, averaged across >4000 frames:


Pyramidal cells in hippocampal CA1 in mice with GCaMP8m
Here, together with Sian Duss, we imaged hippocampal pyramidal cells. Laser power: 35 mW, FOV size: 600 µm, frame rate: 30 Hz , 10x Olympus objective. Unpublished data, averaged across >4000 frames:


A single interneuron in zebrafish olfactory bulb with GCaMP6f
An interneuron recorded in the olfactory bulb of adult zebrafish with transgenically expressed GCaMP6f. Laser power <20 mW, 20x Zeiss objective, galvo-galvo-scanning. (Not shown: simultaneously performed cell-attached recording.) This is from the datasets that I recorded as ground truth for spike inference with deep learning (CASCADE). Zoomed in to a single isolated interneuron, averaged across 1000 frames:

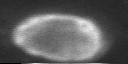
A single neuron in zebrafish telencephalic region aDp with GCaMP6f
A neuron recorded in the telencephalic region “aDp” in adult zebrafish with transgenically expressed GCaMP6f. Laser power <20 mW, 20x Zeiss objective, galvo-galvo-scanning. (Not shown: simultaneously performed cell-attached recording.) This is from the datasets that I recorded as ground truth for spike inference with deep learning (CASCADE). Zoomed in to a single neuron, averaged across 1000 frames:


Population imaging in zebrafish telencephalic region aDp with GCaMP6f
Neurons recorded in the telencephalic region “aDp” in adult zebrafish with transgenically expressed GCaMP6f. Laser power <30 mW, 20x Zeiss objective, frame rate 30 Hz. Unpublished data, averaged across >1500 frames:


Sparsely labeled neurons in the zebrafish olfactory bulb with GCaMP5
Still in love with this brain region, the olfactory bulb. Here with sparse labeling of mostly mitral cells with GCaMP5 in adult zebrafish. This is one out of 8 simultaneously imaged planes, each imaged at 3.75 Hz, with this multi-plane scanning microscope. From our study where we showed stability of olfactory bulb representations of odorants (as opposed to drifting representations in the olfactory cortex homolog), averaged across 200 frames:


Population imaging in zebrafish telencephalic region pDp with OGB-1
Using an organic dye indicator (OGB-1), injected in and imaged from the olfactory cortex homolog in adult zebrafish. This is one out of 8 simultaneously imaged planes, imaged at 7.5 Hz each with this multi-plane scanning microscope. OGB-1, different from GECIs like GCaMP, comes with a relatively high baseline and a low ΔF/F response. The small neurons at the top not only look tiny, they are indeed very small (diameter typically 5-6 um). Unpublished data, averaged across 200 frames:


Pyramidal cells in hippocampal CA1 in mice with R-CaMP1.07
These calcium recordings from pyramidal neurons in hippocampal CA1 exhibited non-physiological activity. Laser power: 40 mW, FOV size: 300 µm, 16x Nikon objective, frame rate 30 Hz. From our recent study on pathological micro-waves in hippocampus upon virus injection, averaged across >1500 frames:


Conclusion
I hope you liked the example images! Also, I hope that this comparison across recordings and brain regions will help to normalize expectations about what to get from a single frame from functional calcium imaging. If you are into calcium imaging, you have to learn to love the shot noise!
And you have to learn to understand the power of averaging to be able to judge your image quality. Only averaging can truly reveal the quality of the recorded images. If the image remains blurry after averaging thousands of frames, then the microscope can indeed not resolve the structures. However, if the structures come out very clearly after averaging, the microscope’s resolution (and the optical access) are most likely good, and only the low amount of photons is stopping you from seeing signals clearly in single frames (which is often, as this gallery demonstrates, not even necessary).

Pingback: There is no recipe for discoveries | A blog about neurophysiology
I am building a Two-photon setup in our lab. The system failed to record any signal. However, I did several checks in terms of alignment and controlling the imaging system. I am wonding if you could give me any suggetion in this situation. Thank you!
That’s a very broad question. If you want useful feedback, it would be good to write what tests of the setup you have done and what the results were. But maybe I can give you some pointers, with which you can then continue to work:
First, I would check that the detection system works. To this end, you turn the PMT gain to a very low value and then, during imaging, shine with the light of your cell phone into the setup. If you don’t see anything for this extremely strong input (be careful not to destroy the PMT via too much light!), then you can be sure that you need to fix the detection pathway (optics or the cabling of PMT to preamplifer or preamplifier to DAQ board).
If the detection works well, check that there is any light coming through the objective when you’re imaging. You can do this using a power meter, or by using a camera that detects infrared. Or you tune the laser (if it’s tunable) to visible spectrum and then see with your eyes (be careful about laser beams and your eyes, I hope you are familiar with the general rules!) whether there is any light going through the objective.
If both detection is fine and light passes through the objective, it is time to use a sample that is as bright as possible. Don’t use a mouse for that, because expression might be absent or weak. use a plastic fluorescent slide for that. Then it will be easy to find the sample and to see something even if the alignment is not good.
Once you see something in the microscope, you can iteratively improve the alignment.
Hope that helps already!
Thank you for your elaborate response. I have started with a very basic and simple two-photon setup.
Earlier, I completed the detection checking part, and it was okay. The way was pretty similar like yours. Even the PMT was able to record noise stimulation from the sample plane (I just moved the VR card!).
However, right now I don’t have any plastic fluorescent slides. But, I tried with commercial fluorescent sample slide F24630 (mouse kidney section with Alexa Fluor™ 488 WGA) and F36909 (Fluorescence Microscope Test Slide #1, for alignment, intensity, & calibration) . But I just ended up recording noise with no fluorescence signal.
Any idea in this current scenario?
I have been following your blog since I started working, and that was a great help indeed! Thanks again!
Hi,
Glad that the blog could help you a bit!
The commercial fluorescent sample slide should work equally well in theory (unless it is very old and bleached out). If you have any kind of other fluorescent microscope in the building, maybe you can check that the slides are fluorescent and not bleached out.
In practice, it is quite easy to create a sample yourself, because most things are fluorescent to some extent. For example, you can just use any kind of colored plastic as sample. It’s often bright as hell, much more than any biological sample.
Have you tried to see whether the laser light passes through the objective?
Good luck with your experiments!
Yes! The laser light passes through the objective. I confirmed it by measuring the power and viewing it using IR detection gun.
Thank you for the ideas and inspiration.
check your emission filters and dichorics are all installed in the right order and the correct way around.
To make it even simpler, you can, to get started, also remove all emission filters except for the IR blocking filter.
Hello! I am back. Thank you for your responses.
I did check the passing of laser rays through the objective lens and measured the power of the beam. Then, I checked the PMT, installation of emission filters and dichorics, which were okay. Finally, I removed the emission filters. Unfortunately, I failed to record any fluorescence signals.
A few days ago, I tested commercial fluorescent sample slide F24630 (mouse kidney section with Alexa Fluor™ 488 WGA) with commercial Confocal, and it provided wonderful images. Then, I used the same sample for two-photon microscopy imaging testing, however, there was no signal.
Do you have any idea how to explain the current situation? Is it the sample? I would like to be confirmed before abadon this biological sample.
I appreciate your efforts to be with me in this frustrating situation. Thanks again!
Hi,
Sorry to hear about these problems!
I have two ideas for reasons why it still does not work for you:
I hope these hints will be useful for you for debugging! If you are then still struggling to see any light, I would really reach out to somebody local close to your lab who could have a look at your system for 1-2 hours. It is very difficult to find the problem if you’re not at the setup.
However, if you don’t find anybody close to your lab who would be able and willing to help you, you can also reach out to me via email; maybe we could find ideas on how to solve your problem when talking.
I did check the alignment for hundred times but yes, it could have problems. I am using an ALCOR femtosecond fiber laser at 920 nm.
If the manual is required, please let me know, I will send it to you via e-mail. I will let you know the result of imaging after checking with brighter sample.
Thank you for your suggetion and kindness.
Why I can’t post my comment!
Sorry, you were blocked by the spam filter of the blog. Only when I saw your comment from July 31st, I checked the spam filter and discovered your messages. Sorry for that, which was unfortunately beyond my control.
Thank you. Now it seems that the comment section is overflooded with my comments. My apology!
My guess is that the ALCOR laser is fairly recent (I think this laser is available since roughly 5 years or so), so I don’t think its age is an issue. Also, I haven’t used this specific fiber laser, so I cannot help with that. I would say that it is very likely that the laser is indeed pulsing. Maybe in your model there is a sync signal (e.g., from a BNC output) that you can plug into an oscilloscope to see whether the laser is pulsing or. I would call the company directly to ask whether this could be an issue and how you can see whether the laser is pulsing or not. This said, I think that this is rather unlikely to be the problem with your setup, and only if you run out of other options, I would check it. But this is something which happened to me several times (although for other laser types).
You mentioned that you checked the alignment hundred times. Did you also check the alignment of the detection path? It can happen that the dichroic mirror is slight rotated, such that all fluorescent light goes through the detection path but slightly misses the PMT detector surface.
Then, I hope that a brighter sample will give you some insights as well.
testing
One comment i would give is that the alcor should come with dispersion compensation unit and you might start with a very bright sample and tune the dispersion compensation for highest SNR (that should mean the dispersion introduced by your microscope is compensated).
Yes, that’s also a good point. It’s very convenient that the alcor laser (and basically all other fixed-wavelength lasers, as ar as I know) has this built-in precompensation unit.
Given your “testing” comment, maybe you were wondering why your comment did not display: The reason for this is that I have to manually clear each and every comment, in order to prevent spam messages. That’s why it usually takes 1-3 days until a comment appears online. I think that’s still acceptable, although of course not ideal :-)
I was able to successfully record photon emission with a brighter fluorescence sample. Now, I am moving forward to the next steps for imaging in our system, also. I would like to thank you for your consistent support. It feels like this challenge opened a door for learning.
This sounds promising! I hope you’ll make progress also with the next steps. And it’s great to hear that you can see this challenge as an opportunity for learning – I really like this mindset!
Thank you for sharing! I’ve learned a lot from your insights. I have a basic question regarding calcium imaging for noisy data. After completing my recording (using a 10X objective, Olympus 2-photon microscopy, GCAMP8m, 40mW, 30Hz), I used Suite2p to extract raw data and calculate ΔF/F. However, I noticed that the ΔF/F signal has a lot of noise. Do you have any suggestions or pipelines for addressing this issue? The noisy data is affecting the Cascade results, which are not ideal.
Thank you for your comment! As I wrote in the blog post, high noise levels may come from several sources, e.g., low expression, low excitation efficiency, a suboptimal fluorescence detection beam path, or imaging in deep brain areas with high losses due to scattering. In many cases, these issues cannot be fully circumvented (or are difficult to figure out). In this case, you might consider using denoising approaches such as DeepCAD.
However, if your raw data look already good while the data extracted via Suite2p look noisy, this can have several reasons that you might want to check. (1) Some ROIs in Suite2p are too small, only covering a fraction of a neuron. Therefore, the signal integrated over this ROI will be weaker and the noise higher. A solution would be to discard these noisy ROIs (if only a subset of these ROIs is affected). (2) Sometimes there is an issue with the PMT offset or another offset value, resulting in a division by something close to zero by Suite2p, resulting in turn in high apparent noise levels. Maybe you can check for this. Do the noise levels look similar when you extract a raw fluorescent trace in ImageJ or FIJI?
Finally, in principle, Cascade should be quite good at dealing with noisy data. If you find that this does not work out in your case, it would be helpful to reach out while sharing how your data look like. You can do this via email (p.t.r.rupprecht+cascade@gmail.com) or by opening an issue (https://github.com/HelmchenLabSoftware/Cascade/issues). With a clearer idea about your data, I would be able to help you better with suggestions to address your issue. There is no solution that fits all use cases.