In point-scanning microscopy like two-photon or confocal microscopy, a focused laser beam is scanned across the field of view and thereby sequentially recovers an image of the object. In this blog post, I will discuss the idea that scanning faster across the field of view would increase the total amount of collected fluorescence. This idea is based on the experimental finding that high-intensity laser light could induce long-lived and non-fluorescent triplet states of the fluorophore molecule while scanning the sample; when the laser is scanned only slowly across the sample, it would therefore try to image fluorophores that are already in their “dark” triplet states. Imaging dark fluorophores would result in an overall decreased fluorescence yield. I tested this hypothesis directly with resonant scanning two-photon microscopy in the living brain tissue together with typical fluorophores (GCaMP6f, OGB-1). The main result from these experiments is that I could not find a substantial effect of the triplet states under these realistic conditions, and therefore no advantage in terms of fluorescence yield gained by an increased scan speed.
Introduction
Fluorophores are molecules that can re-emit light after absorbing a photon themselves. This effect of fluorescence can be described as a state change of the fluorophore from ground state to an excited state upon absorption of the incoming photon, and as a state change from an excited state back to the ground state, together with emission of the outgoing photon (see this Wikipedia article). The lifetime of the fluorescent state is often a few nanoseconds. However, there are often additional excited states that are more long-lived, such as so-called triplet states. The transition probabilities from ground state to triplet state (and vice versa) are very low due to the exclusion principle that derives from quantum spin mechanics. The lower transition probabilities make these states less likely to occur during fluorescence microscopy but also render the triplet state longer-lived once it is attained. These long-lived triplet states could have rather undesired consequences, since a fluorophore in the triplet state is unable to absorb or emit photons, therefore becoming “dark” or non-functional from the experimenter’s perspective.
When triplet states play a prominent role in laser-scanning microscopy, one can avoid their detrimental effects by simply scanning faster. Scanning slowly across the sample means that the same fluorophores are hit with light over and over within a short time window. In such a scenario, some of the fluorophores are already in a dark triplet states, resulting in lower overall fluorescence. Faster scanning avoids this problem, and it has been shown for confocal microscopy (Borlinghaus, 2006) as well as STED microscopy (Schneider et al., 2015; Wu et al., 2015) that a large signal increase can be achieved simply by faster scanning.
For two-photon microscopy, the situation is less clear. It has been shown that scanning at all compared to non-scanning two-photon fluorescence correlation spectroscopy results in higher photon yield (Petrášek and Schwille, 2008), but this specific fluorescence configuration cannot be translated easily to two-photon imaging used by neuroscientists. However, there is one publication that demonstrated that microsecond-long triplet states do play a large role for two-photon microscopy (Donnert et al., 2007). This paper has been cited as a standard reference to justify the advantages of fast scanning approaches (e.g., Chen et al., 2011), and some two-photon microscopy approaches directly designed their scanning approaches to reduce triplet-induced reduction of fluorescence yield (Gautam et al., 2015; Castanares et al., 2016; Karpf et al., 2020). However, another study showed an effect that seemed to be contradictory to the Donnert et al. results (Ji et al., 2008), and the interpretation of both papers was discussed repeatedly, e.g., by Andrew Hires and on Labrigger. The consensus, if there was any, seemed to be that it probably depends on the sample. The experiments by Donnert et al. had been done with GFP fixed on a coverslip, but one can easily imagine that a fluorescent protein in vivo might behave differently.
So I decided to probe these results with typical samples and procedures used by neuroscientists – using video-rate calcium imaging of neurons in the living brain. I did not do this out of pure curiosity about photophysics, but because of the implications of these photophysics for the design of two-photon imaging modalities. If triplet states were indeed an important factor for two-photon imaging of such samples as suggested by Donnert et al., a slightly modified scanning scheme (or parallelized scanning schemes, as reviewed by Weissenburger and Vaziri, 2018, Figure 3) or adapted laser repetition rates would have huge benefits for the total fluorescence yield.
The main finding of the Donnert et al. paper was the induction of dark triplet states by the scanning laser, and a key prediction was that faster scanning would decrease the probability that an imaged fluorophore is in a dark state; in short, that faster scanner would increase fluorescence yield. Therefore, I wanted to systematically understand whether fluorescence for two-photon microscopy depends on scan speed. To study this dependence experimentally in a typical in vivo sample , I used a resonant scanning microscope. For a resonant scanning microscope, the physical scan speed across the sample can be adjusted by changing the scan amplitude (often called the Zoom setting). High zoom would result in low scan speed across the sample, while low zoom would result in high scan speed, enabling a simple characterization of scan speed versus fluorescence yield. According to Donnert et al., higher scan speed would result in higher fluorescence yield compared to slower scan speed when imaging the same spot.
Experimental approach
For a resonant scanning microscope, the scan speed can be derived from the position x(t) of the laser beam focus:

with the resonant frequency (f = 8 kHz) and the amplitude (x0 = 500 μm), which determines roughly the size of the field of view (FOV, here: 1000 μm). The scan speed v(t) is the derivative of the position x(t) with respect to time and therefore also follows a sinusoidal trajectory that reaches its peak in the center of the FOV:

with the speed at the center of the FOV given by

For higher zoom values (i.e., a smaller FOV), the value is reduced by the same factor (at least in the microscopy software I used for these experiments!). Thereby, it is possible to test a wide range of scan speeds just be changing the zoom setting of the resonant scanner. Since the resonant scanner can span zoom levels between 1x to 20x (for much higher zoom settings, a resonant scanner can become unstable due to the small scan amplitude), a range of speeds between 25 μm/μs and 1.25 μm/μs is spanned.
This speed should be compared to the lateral resolution of the microscope, which is for the microscope used in this experiment around 0.4 μm FWHM (full width at half maximum of the point spread function). To scan over this resolution-limited spot, the resonant scanner needs 0.016 – 0.32 μs. For a pulsed laser with a repetition rate of 80 MHz with 12.5 ns between two pulses, this corresponds to 1.3 – 26 pulses per resolution-limited spot. Let’s call this number the cumulative pulse count per resolution-limited spot (CPC) from now on.
Intuitively, this means that each time a laser pulse tries to excite a fluorophore, this fluorophore has very recently already been hit by a number n of previous pulses, approximately n = CPC/2. If there is a chance that a pulse drives the fluorophore into a dark state that makes the fluorophore non-excitable for a handful of microseconds (e.g., a triplet state), then the CPC can be used to calculate the number of fluorophores remaining to be excited:

λ is a constant that depends on the applied laser intensity and the fluorophore itself. More concretely, the constant λ is the event rate of a fluorophore going into the dark state. The exponential decay with CPC should hold true as long as these events occur within a time window that is shorter than the recovery timescale of the dark state (for triplet states, this is ca. 1 μs). The main consequence is that the fluorescence yield is obviously proportional to the remaining number of excitable fluorophores, Nremaining.
Therefore, if these events that generate dark triplet states were very unlikely (e.g., λ ~ 500), the effect of fast scanning would not really make a difference. However, if λ was ~1, the effects would be dramatic. How can we distinguish these scenarios?
Experimental implementation, part I
With a resonant scanning microscope, the experiment is easy to perform for a dye in a solution. You simply have to generate a homogeneous sample and image first with high zoom and low zoom, then compare the fluorescence in the central region where the scan speed is maximal.
Unfortunately, biological samples are not homogeneous, and also not stationary in the case of calcium indicators in living neurons. To image in a biological but still rather homogeneous sample, I used a transgenic GCaMP6f zebrafish line. I imaged in an explant of a dorsal forebrain region that I knew was labeled very densely and very homogeneously (described by Huang et al., 2020). But the neuronal somata with the nice nuclear exclusion of GCaMP generated a lot of undesired variability.
The solution to circumvent these sources of variability is systematic averaging. In a first approach, I took advantage of the fact that I had written large parts of the microscopy control software myself. I wrote a helper program that performed continuous imaging but randomly moved the stage to different positions every few seconds, thereby averaging across all these inhomogeneously labeled FOVs:
After a minute, the program automatically changed the Zoom setting to a different, random value and recorded again the video with intermittent stage movements:
To see an effect, I performed these experiments with rather high laser power (50-80 mW below the objective) and at rather low wavelengths (typically 800 nm, which had been used previously by Donnert et al.).
In the following plot, each data point corresponds to one movie as shown above. Keep in mind that a zoom setting corresponds to a specific CPC. For example, a zoom setting of 1 corresponds to a CPC of 1.3. However, I could not see any dependence of the total fluorescence on the cumulative pulse count CPC (left), so it was not worth determining a numeric value for λ. Interestingly, the total fluorescence decreased slightly but clearly visible over time (right), but since the zoom setting sequence was randomized, this did not reflect any dependence on the CPC:
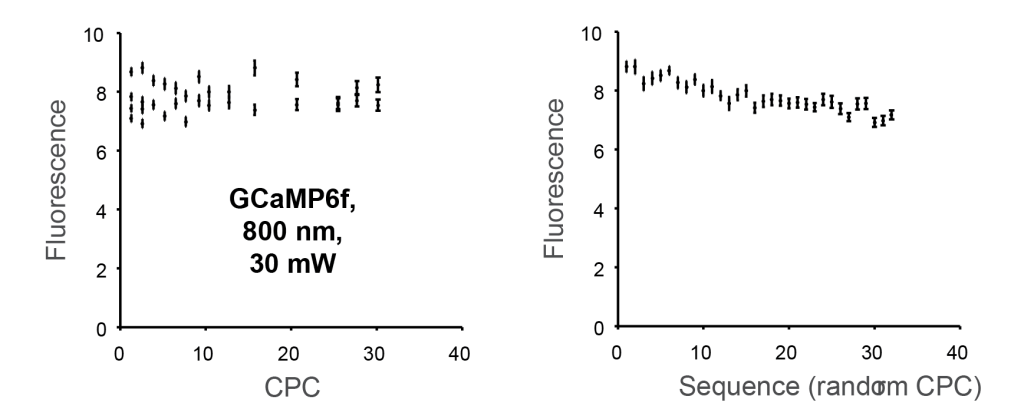
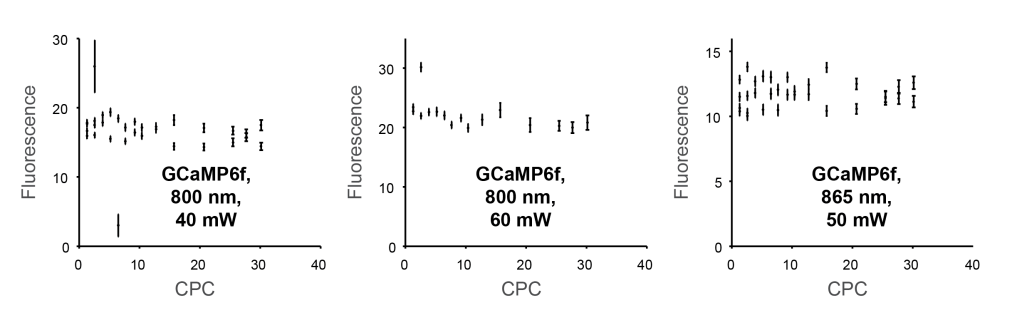
This finding also held true for repetitions of the same experiment at different locations of the fish brain and with slightly changed wavelength or average power:
Next, I performed the same experiment with a different fluorophore. I injected OGB-1 into the homolog of piriform cortex of zebrafish. This is a large and relatively uniform region in the zebrafish forebrain that allows the dye to diffuse more or less homogeneously, at least if you’re lucky. When I analyzed the experiments, I found again no visible effect, no matter the laser power or the laser wavelength:
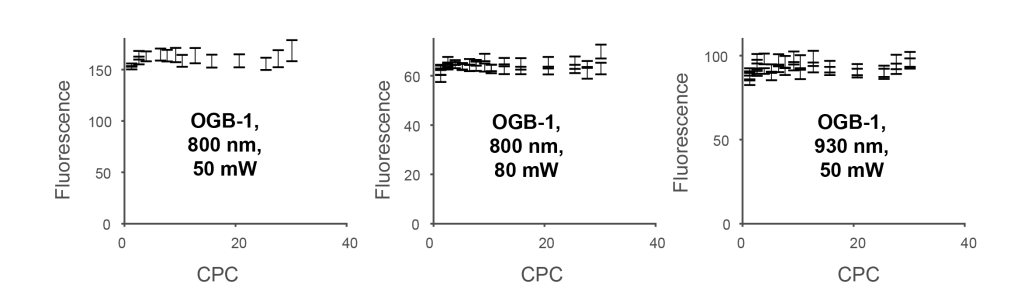
Together, these experiments strongly suggested that there is indeed no effect of triplet states and therefore no benefit of fast scanning to increase the fluorescence yield.
Experimental implementation, part II
However, to convince myself more about this experimental finding that seemed to be at odds with my expectations from the Donnert et al. paper, I used a second, slightly more direct approach. Instead of comparing sequential recordings at different zoom levels, I thought it would be interesting to record the fluorescence while changing the zoom level continuously. This would enable me to measure the dependence of fluorescence on CPC more quickly and therefore also for a larger set of power settings and wavelengths. To continuously change the zoom setting, I used a DAQ board to generate a low-frequency sine signal that modulated the zoom level from 1 (peak of the sine) to ~20 (trough of the sine) with a period of ca. 13 seconds. (To keep track of the sine signal, I also connected the command signal with the second input channel of the microscope.) That’s how these experiments looked like:
Of course it is important to use not the entire FOV but only the central vertical stripe that remains more or less stationary. I used only a small vertical window of 7 pixels for the analysis. A single experiments resulted in a result such as shown below, plotting CPC (top) and fluorescence (bottom). The fluorescence clearly shows some variability stemming from active neurons (after all, we’re still dealing with a living brain here!):
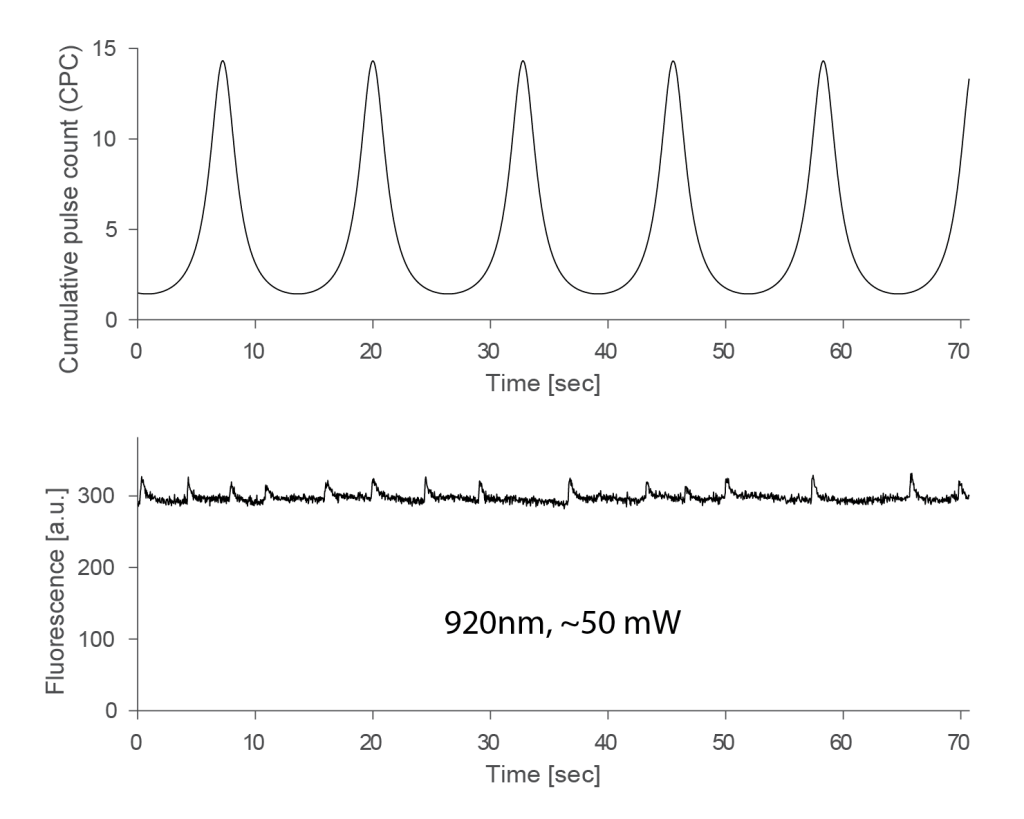
In the plot above, no obvious relationship between CPC and fluorescence can be seen, and when I changed the power at 920 nm between 15 mW and 60 mW (this was the maximum that I could get with this system), I could not see any effect. I therefore show here all experiments performed at 920 nm, pooled across power settings and across two fish (total of ca. 30 recordings, each a few minutes):
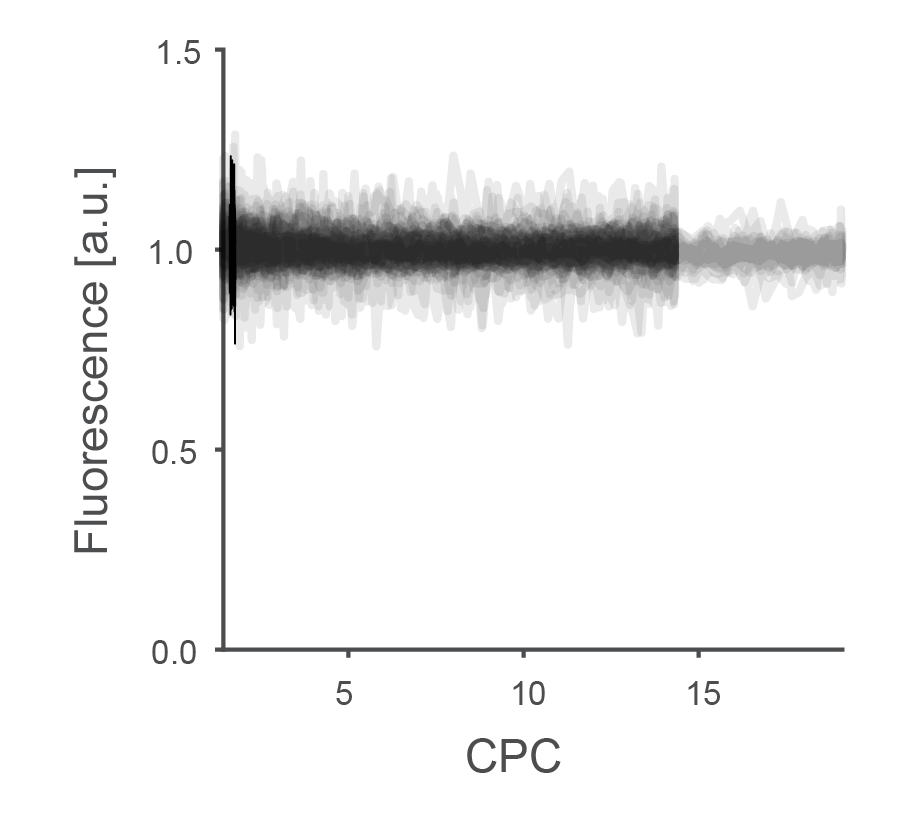
For some experiments, the maximum zoom level was around 14, which I extended for a subset of experiments to something closer to 20.
I performed the same experiment also at 800 nm. I could also increase the laser power, simply because the laser provided more power at this wavelength range. However, these levels cannot go to arbitrary values. At a certain threshold that also depends on the duration of the exposure, all neurons across the zebrafish’s brain region become bright, resulting in a wave of high-calcium neurons that propagates through the brain. To avoid this, I used a maximum power of 75 mW at 800 nm. The result, again pooled across laser powers between 40 and 75 mW, showed an effect, albeit a bit subtle:
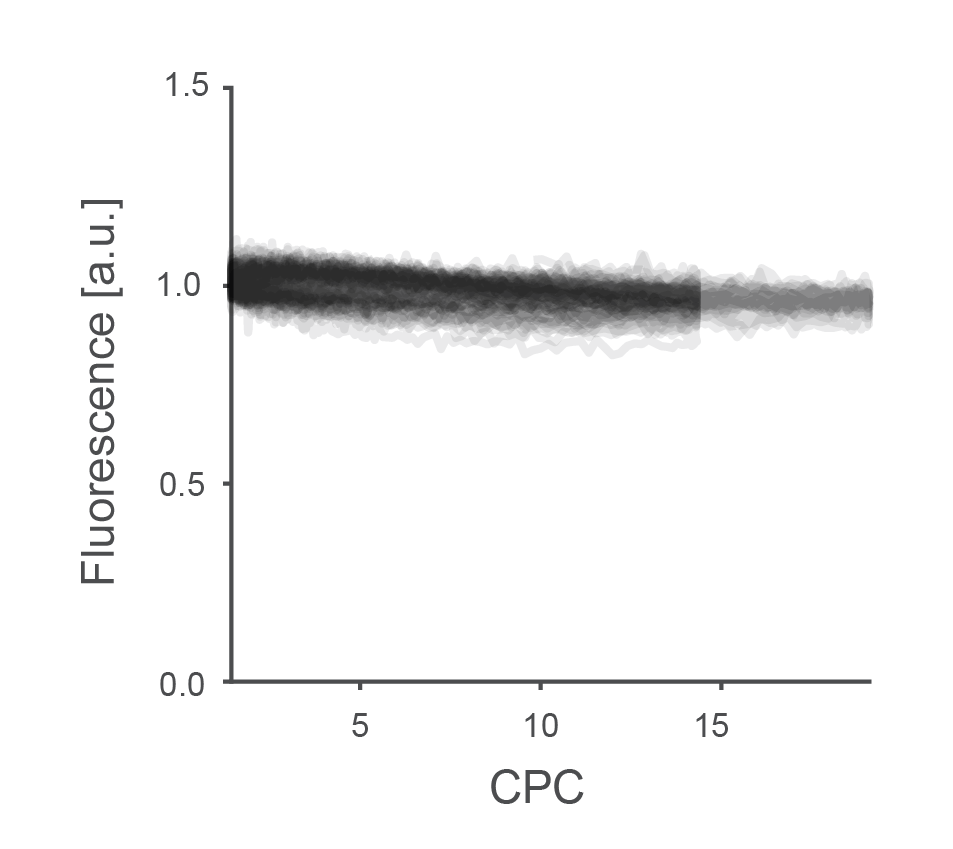
Fluorescence was indeed slightly higher for very low CPCs. However, the effect was much smaller than expected from experiments in vitro by Donnert et al.. Overall, such a small effect which, in addition, only appeared at the less relevant wavelength of 800 nm, seemed of little relevance for practical purposes.
Therefore, with fluorescence depending on CPC only in minor ways and under non-typical imaging conditions, the suggested triplet states seem to be not relevant for in vivo calcium imaging situations; and as a consequence, one would be ill-guided to assume that faster scanning yields higher fluorescence yield for two-photon microscopy by avoiding these μs-long dark states.
As a side-note, when I tried to analyze the power-dependency of this weak effect observed at 800 nm, I came across a weird effect. There was indeed some sort of increased fluorescence at lower zoom levels. However, this increase came with a delay of several seconds during the above experiments, resulting in a hysteresis:
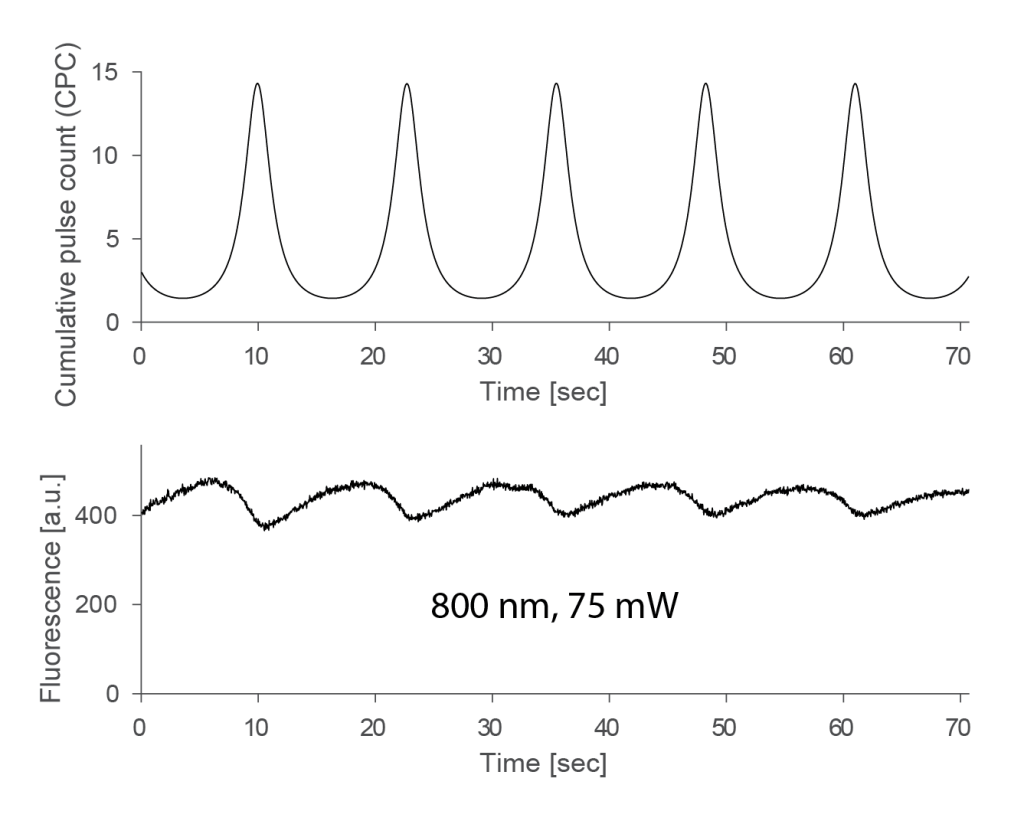
Due to this longer time-scale, this effect has nothing to do with the short-lived μs-triplet states but is something different entirely. Photophysics is really complicated! This additional observation made me stop experiments, because I realized that these things would be more difficult to figure out, on top of a very small and probably irrelevant triplet effect.
Conclusion
I did not find any evidence for a substantial effect of triplet states during typical conditions (calcium imaging of neurons with a Ti:Sa laser and <100 mW power at the sample). I therefore do not see a benefit in terms of fluorescence yield by faster scanning. The triplet states on the timescale of few microseconds that had been observed by Donnert et al. for fixed samples do not seem to play a major role under the investigated conditions. It is always challenging to convincingly show the absence of an effect, but my experimental results convinced me to not further investigate fast scanning and multiplexing schemes as a means to increase fluorescence yield.
[Update August 2021: Christian Wilms pointed out that the most obvious difference between my experiments and the Donnert et al. study is that the dye molecules were freely diffusible in my experiments but fixed in the Donnert et al. experiments. Consistent with that, he also noticed that the paper from Ji et al., which found results contradictory to the Donnert et al. paper, was mostly based on experiments with freely diffusible dyes, while STED experiments, which clearly showed the effect, are mostly based on fixed fluorophores.]
Multiplexing or other modified distributions of excitation photons in time and space might still be able to increase fluorescence yield for certain fluorophores and conditions, but probably not under conditions similar to the ones I investigated.
The experiments described above are not fully systematic and cover only a specific parameter regime of excitation wavelengths, fluorophores and laser powers. However, anybody with a resonantly scanning two-photon microscope can easily reproduce these findings for any other scenario. Simply switch between high and low zoom settings and check whether the brightness in the center of the FOV changed substantially or not. Quantification of possible effects requires careful averaging, but a quick and qualitative confirmation or refutation of the above findings would be very easy to do for any experimenter.
Acknowledgements
The experiments described above were carried out in the lab of Rainer Friedrich at the FMI in Basel. I’m thankful to Christian Wilms, who encouraged me to analyze and write up these experiments after a discussion on Twitter.

Pingback: Interesting papers on recent neuroscience methods | A blog about neurophysiology